|
|
|
Purpose This tutorial is designed to illustrate the application of NMFF and the MMTSB Tool Set to the flexible refinement of an atomic level structure into an electron density map from cryo-EM, electron tomography, etc. In this exercise we will create an electron density map corresponding to the closed structure of adenylate kinase (right figure) and refine the atomic coordinates of the open conformation (right figure) into this density using the normal mode flexible fitting algorithm. To perform the calculations in this tutorial you will need to have the NMFF suite of programs installed. Installation instructions for NMFF can be found here. We will also use parts of the MMTSB Tool Set. Other useful programs include vmd, which is used to visualize the structural and electron density data, however, your favorite structure/density viewing/manipulation program (e.g., O, etc.) can also be used. |
 |
|
Preparing the data First we need to get the "raw" pdb files for the closed (1AKE.pdb) and open (4AKE.pdb) states of adenylate kinase. These can be obtained from the pdb by or directly downloading them using the links above. We want to extract the coordinates from chain A for each system and center the coordinates before computing the electron density map. To do this, for our target molecule 1ake, we use the MMTSB Tool convpdb.pl. The following unix command will do this (Note , unix> denotes the unix system prompt and the command follows.) unix>convpdb.pl -center -nohetero -chain A 1AKE.pdb > 1ake_aa_center.pdb With this command we created the new pdb file named 1ake_aa_center.pdb. Let's now make the electron density map using the NMFF suite component program pdb2sim_map. This program requires three arguments and takes an optional fourth. To make the electron density map representing 1ake_aa_center.pdb at approximately 10 Å resolution with a 1 Å grid spacing, use the command. unix>pdb2sim_map 1ake_aa_center.pdb 5 1ake_aa_center.xpl 1 |
|
Note that the electron density map is written in the X-plor format. Also, see that the 10 Å resolution is achieved by setting the 2nd parameter to 5 (desired resolution/2). The computed electron density is shown enveloping the cartoon structure of the closed form of adenylate kinase in the figure on the right. Next we will superpose chain A from the structure 4AKE.pdb onto the backbone of 1ake_aa_center.pdb and then rotate this conformation by 20° around the x-axis to "misalign" the structure to be refined and the density, for illustrative purposes, using a combination of MMTSB Tools and unix pipes. The tools are convpdb.pl and lsqfit.pl. The command is: unix>convpdb.pl -chain A -nohetero 4AKE.pdb | lsqfit.pl -sel ca 1ake_aa_center.pdb | convpdb.pl -rotatex 20 > 4ake_aa_rotx20.pdb |

|
|
Anticipated output and further analysis Some snippets of output from running NMFF on this refinement program are as follows:
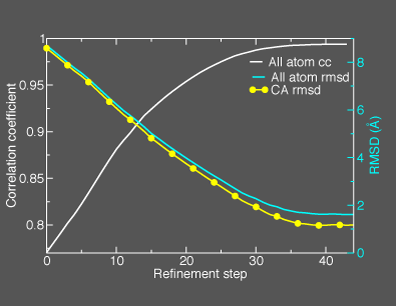
This general information flow goes on for (in this case) 44 iterations. Note that the initial correlation coefficient is about 0.76 and after the last iteration the refinement exits after making an attempt to improve the correlation coefficient further (now at 0.99) with a newton-raphson refinement step. In the figure below we illustrate the course of the refinement by plotting the (left axis) correlation coefficient between the model computed density and the target electron density 1ake_aa_center.xpl versus refinement step and (right axis) the Ca and all-atom root-mean-square deviation between the model being refined with NMFF (starting from 4ake_aa_rotx20.pdb) and the target closed-state structure (1ake_aa_center.pdb). In the animation to the right, we can see the course of the motion of the 4ake-based model into the density from 1ake. |
|
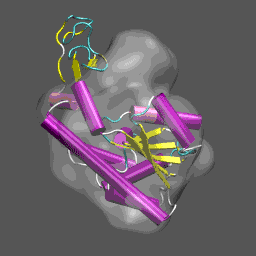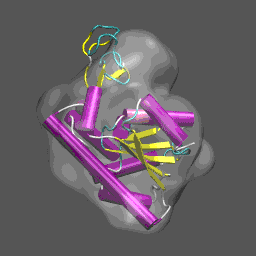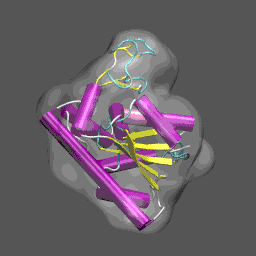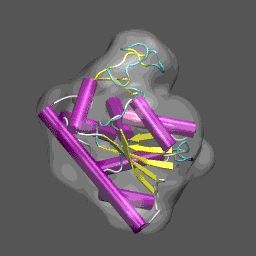 |