NMFF Virus Capsid Refinement Tutorial
|
|
Purpose
This tutorial is designed to illustrate the application of NMFF and the MMTSB Tool Set
to the flexible refinement of an atomic level structure of a viral capsid into an electron density map
from cryo-EM,
electron tomography, etc. In this exercise we will create an electron density map corresponding
to the swollen state of STMV (right figure) and refine the atomic coordinates of
the native STMV conformation into this density using the normal mode flexible fitting
algorithm. Models for the native and "swollen" STMV are shown in the figure on the right and below. We note that
STMV has not been characterized in a swollen state, as can be observed for a number of other viral capsids. However, we
are using this artificial example because of the small size of the capsid protein and the STMV viral capsid.
STMV, or Satellite Tobacco Mosaic Virus is a small T=1 viral capsid. You can learn more about
STMV
at the ViperDB
(VIrus Particle ExploreR DataBase)
To perform the calculations in this tutorial you will need to have the
NMFF suite of programs
installed. Installation instructions for NMFF can be found
here.
We will also use parts of the
MMTSB Tool Set.
Other useful programs include vmd,
which is used to visualize the structural and electron density data, however, your favorite structure/density
viewing/manipulation program (e.g., O, etc.) can also be used.
|
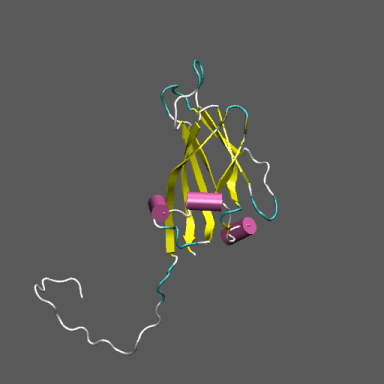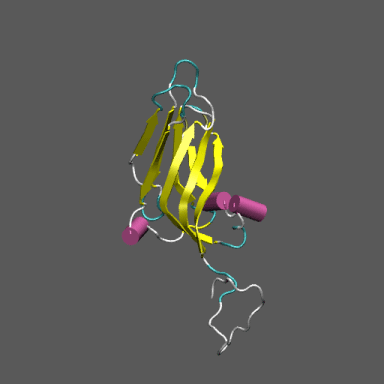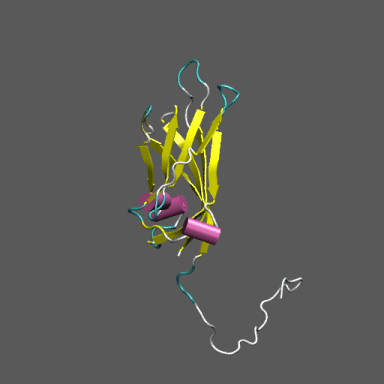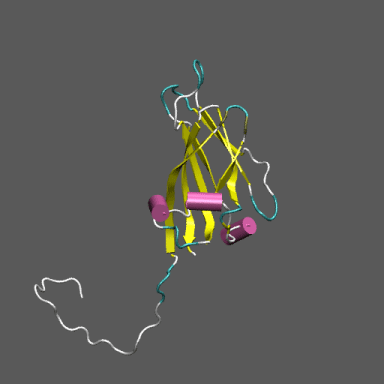
Satellite Tobacco Mosaic Virus
(capsid protein)
|
Preparing the data
First we need to get the "raw" pdb files for the capsid protein in the native state and construct the entire
capsid. To do this we can go to the ViperDB entry for STMV
and download the pre-constructed coordinates for the full capsid by
clicking on the download full capsid (in the middle panel). This file, called 1a34_full.vdb, will be
used to construct the native CA based model we refine into the "swollen" density for STMV. The model structure we will use to build the
swollen density from is 1a34_swollen_ca.pdb
(Note this is a model constructed by expanding the native conformation along its icosahedral
normal modes to yield a conformation approximately 20 Å expanded from the native. It is not a physically realized conformation.)
Download these files using the links above.
|
|
We now want to extract the CA atoms from the structure 1a34_full.vdb. To do this it's easiest to use the unix command grep.
The following unix command will do this (Note , unix> denotes the unix
system prompt and the command follows.)
unix>grep CA 1a34_full.vdb > 1a34_i60_ca.pdb
With this command we created the new pdb file named 1a34_i60_ca.pdb.
Let's now make the electron density map for the swollen state using the NMFF suite component program pdb2sim_map.
This program requires three arguments and takes an optional fourth. To make the electron density map
representing 1a34_swollen_ca.pdb at approximately 24 Å resolution with a 2 Å grid spacing,
use the command.
unix>pdb2sim_map 1a34_swollen_ca.pdb 12 STMV_swollen_24.xpl 2
Note that the electron density map is written in the X-plor format. Also, see that the 24 Å resolution is
achieved by setting the 2nd parameter to 12 (desired resolution/2).
|
 |
We are now ready to move on to running the refinement. Note that you can skip these steps and directly
download the density STMV_swollen_24.xpl and the coordinates
1a34_swollen_ca.pdb
and 1a34_i60_ca.pdb.
Refining STMV with NMFF
The control program for running NMFF is the perl script nmffem.pl. This script reads the input
parameters that controls the refinement as well as runs the refinement through calls to component programs
that are part of the NMFF suite. The input file is called nmff.inp and contains the following lines:
nmff.inp
original pdb = ../structure/1a34_i60_ca.pdb
target map = ../density/STMV_swollen_24.xpl
maximum displacement = 1.0
maximum number of modes = 15
cutoff = 8.0
nmff directory = ../../../src/nmff/em
nma directory = ../../../src/nma/rtb
resolution = 24.0
T number = 1
In nmff.inp, as shown above, original pdb
corresponds to the atomic structure that is used to start the refinement,
target map
corresponds to the electron density map that is the target of the refinement,
maximum displacement
is the variable, in Å, that controls the size of step taken along any normal mode to displace atomic positions in
adjusting the model structure to maximize the correlation between the model and the electron density.
NMFF uses a small set of normal modes (of which there are 3N-6, where N is the number of atoms or sites) as the coordinates
to move the atomic positions to better fit the target electron density. Generally
the first 6 normal modes describe overall translations and rotations of molecule. If we optimally aligned the structure
into the density prior to starting the refinement, e.g., using Situs
and rigid-body translations/rotations, then it is probably not necessary to use these modes during the NMFF
stage. Also, in symmetric structures like an icosahedral virus capsid, only certain modes (those obeying the same symmetry as
the underlying models - EM density or atomic structure) will move the structure towards its target. These (icosahedral) modes
are identified by NMFF and chosen for the directions of displacing the atomic model. The line
maximum number of modes is the specification of the last
mode to use. Thus for this NMFF refinement we use a maximum number of 15 icosahedral modes; that is,
we search a 15 dimensional variable space to flexibly fit the structure to the target density.
The line beginning with cutoff specifies the distance cutoff to be used
in constructing the elastic network model for normal mode analysis. For CA atom
refinements, such as we are doing here, a cutoff of 8Å is appropriate. The next two lines indicate where the
NMFF electron density map related (nmff) and normal mode analysis (nma) related programs reside. If these were installed
in a specific directory,
e.g., by specifying BINDIR= when you configured and installed NMFF, that path goes here. Finally, we specify
the approximate resolution of the electron density map to which we are flexibly fitting the atomic structure; in the present
example the resolution is 24 Å and the
T number for the viral capsid - 1 in this case.
We are now ready to run NMFF and refine the native conformation of STMV into the electron density of the swollen state.
To do this the unix command given below accomplishes this.
unix>nmffem_virus.pl < nmff.inp
The refinement from this starting configuration, with the all atom model, takes about 45 minutes on an
Intel PIV (3GHz) machine.
|
Anticipated output and further analysis
Some snippets of output from running NMFF on this refinement program are as follows:
nmff-em start at 16:20:18
nmff-em parameters
initial pdb structure = ../structure/1a34_i60_ca.pdb
number of atoms in the pdb file = 8820
target em map = ../density/STMV_swollen_24.xpl
resolution of the cryo-em map = 24.0
cut-off used in ennma = 8.0
lowest frequency mode included = 1
maximum number of mode used for the refinement = 15
number of iteration loops = 300
virus T number = 1
maximum displacement allowed at each step (A) = 1.0
maximum number of trial of gradient = 2
nmff directory = ../../../brooks/src/nmff/em
nma directory = ../../../brooks/src/nma/rtb
correlation coefficient output = corr_coef.dat
initial cc: 0.849644
begin iteration at 16:20:28
-----------------------------------------------------------------------
begin iteration 0 with the modes 1 - 5 at 16:20:28
open current pdb file: mov0.pdb
start normal mode calculation at 16:20:28
file# mode#
1 95
2 24
3 180
4 35
5 135
start gradient calculation at 16:21:00
calculate gradients for modes 1 - 5
start structure deformation at 16:23:59
atom with maximum displacement = 7953
with scale = 206118.815222269, move by 1 A
mode amplitude
2 -74.1522744
1 -11.3035764
3 -10.9114354
4 -0.00176296514
5 -0.000842135521
total rmsd 0.807096505, max displacement 1, at atom 7953
result of iteration: 0, gradient try: 1, cc: 0.876496, saved mov1.pdb
cc increased to 0.876496, previous cc was 0.849644
proceed to next iteration, continue gradient mode
-----------------------------------------------------------------------
-----------------------------------------------------------------------
------------------------DELETED OUTPUT---------------------------------
-----------------------------------------------------------------------
-----------------------------------------------------------------------
-----------------------------------------------------------------------
begin iteration 10 with the modes 1 - 5 at 16:57:32
open current pdb file: mov10.pdb
start normal mode calculation at 16:57:32
file# mode#
1 174
2 36
3 142
4 27
5 110
start gradient calculation at 16:58:04
calculate gradients for modes 1 - 5
start structure deformation at 17:01:02
atom with maximum displacement = 1632
with scale = 5198671.31990427, move by 1 A
mode amplitude
2 -72.7616435
3 19.4446423
1 -1.34066975
4 0.0766861205
5 0.00383251768
total rmsd 0.802077389, max displacement 1, at atom 1632
result of iteration: 10, gradient try: 1, cc: 0.993102, saved mov11.pdb
correlation coefficient decreased
move again along the same modes with amplitude divided by 2
mode amplitude
2 -36.3808218
3 9.72232116
1 -0.670334875
4 0.0383430603
5 0.00191625884
total rmsd 0.401038694, max displacement 0.5, at atom 1632
result of iteration: 10, gradient try: 2, cc: 0.993874, saved mov11.pdb
cc increased to 0.993874, previous cc was 0.993767
proceed to next iteration, start newton-raphson
-----------------------------------------------------------------------
begin iteration 44 with the modes 1 - 26 at 12:27:03
too many normal modes (26) are included (> 20); give up
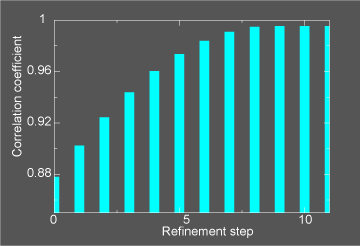
This general information flow goes on for (in this case) 11 iterations. Note that the initial correlation coefficient is about
0.85 and after the last iteration the refinement exits after making an attempt to improve the correlation coefficient
further (now at 0.99) with a newton-raphson refinement step.
In the figure below we illustrate the course of the refinement by plotting the (left axis) correlation coefficient between the
model computed density and the target electron density STMV_swollen_24.xpl versus
refinement step.
In the animation to the right we can see the course of the motion of the 1a34-based model into the density from
the swollen state.
|
|

|
|