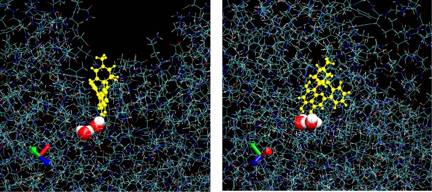
In the input file, mmff_inter_energy.inp,
the MMFF force field is first setup. As MMFF is a Class II force field it requires significantly more input
than the standard CHARMM force fields. The necessary files are in the toppar and toppar/data subdirectories. Once MMFF is setup the inhibitor,
protein and two waters are read and the respective structures generated. This is followed by calculation
of the interaction energy between 1) the drug and the protein and 2) the
drug/water complex and the protein. This is first done using the crystal structure directly, followed by the
minimization of the two waters with the drug and protein constrained. Additional interaction energy commands
are performed following minimization of the drug and water with the protein
fixed and of the drug, water and the local protein environment minimized. The resulting interactions energy are output in the file 1c8k_cpbh_1.inter for
analysis.
When using the INTERaction energy command it is important
that the nonbond lists be setup properly. This requires a call the ENERgy prior to the INTERaction command. Importantly, when using the CONS FIX
command for the minimization it is essential to remove the constraints (CONS
FIX sele none end) then perform an ENERgy command prior to the INTERaction
command. This is necessary due to
the CONS FIX command altering the nonbond list in order to save computer
time. Finally, the INTERaction
energy command may be used with the standard CHARMM force fields in the same
way as with MMFF.