Objective
and Overview
The objective of
this tutorial is to introduce users to
step-by-step procedure to build a membrane system with
CHARMM. In this
tutorial we will:
- Generate the
WALP16 peptide
- Solvate the
peptide with TIP3 water and DMPC lipids
- Minimize and
equilibrate the system
Due to the complexity of lipid molecules, relatively long equilibration
is required after assembly of membrane protein/peptide, lipid
molecules, and water
(sometimes ions).
It
should be stressed
here that the procedure below is not the only way to build a membrane
system.
|
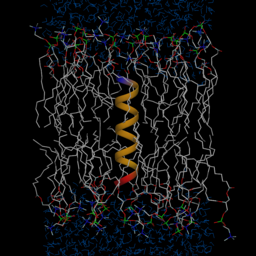
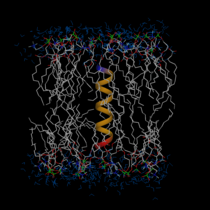
A molecular graphical
view of the WALP16
peptide embedded in a DMPC membrane in an aqueous solution.
|
Step-by-Step for building a membrane
system
| pept.inp |
This
is a
step for preparing and orienting the membrane protein/peptide of your
interest. In this example, a helical
conformation of the transmembrane WALP16 peptide is generated using its
sequence information (GWWLA LALAL ALAWW A). The helical principal axis
is oriented along Z.
|
| step1.inp |
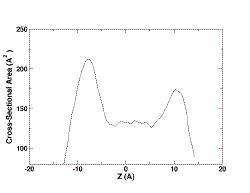
To
determine the system size or number of lipid molecules, one
has to calculate the cross-sectional area of the protein/peptide along
Z. In this example, if you plot "step1.plo", the result of the input
file, you will see
Note that the asymmetry in the profile results from Leu-Ala alternation
in the sequence. For the present illustration, we will simply take the
average of upper and lower minima and save it in a file called
"step1.str" for the next step.
|
| step2.inp |
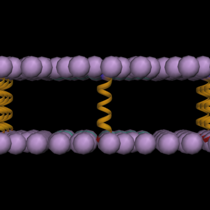
This
step is to build optimal positions of pseudo-lipid big spheres, which
will be used later to place real lipid molecules. In this step, we have
to determine the size of the system along XY directions.
60.7
A**2 / DMPC at 303 K
64.0
A**2 / DPPC at 323 K
In this example, we will use 16 DMPC lipids for each leaflet (upper and
lower to form a bilayer). Therefore, the system size can be determined
from the area of both lipids and the WALP peptide. In the input,
calc
BoxsizeXY = sqrt ( @Nlipid / 2.0 * 60.7 + @PeptArea )
At the end of the calculation, "step2_img.pdb" will look like these;
Note that the magenta
spheres are in the primary system and the rest is in images along XY.
|
| step3.inp |
We
now can put lipid molecules to each position of the big sphere by
randomly picking a lipid conformation from a library of DMPC. Note that
the DMPC lipids in the library contain some water molecules around the
head group. One may see quite some of overlaps between lipid molecules
due to the random picking.
|
| step4.inp |
Now,
it is time to add some water molecules to fully solvate the system.
Here, we will use a water box with a length of 22 A. One should
determine an appropriate size based on the protein size. By starting
the box from +/- 12 A along Z, we have a system size of 68 A along Z;
calc
BoxsizeZ = ( 12.0 + @waterlength ) * 2.0
Once we remove all the water molecules close to the previous components
(peptide, lipids, and solvation water) within 2.6, we separate water
and lipids into different files ("step4_dmpc.crd" and "step4_tip3.crd").
|
| step5.inp |
So
far we
just built the system by putting the components piece by piece, and
thus each
component is totally uncorrelated. After assembly in this step, we
will minimize and equilibrate the system in NVT ensemble with
restraint potentials (restraint.str)
to place things where they supposed to be initially.
|
| equi1.inp |
We
continue
and continue the system equilibration in CTPA ensemble with PME
(Particle
Mesh Ewald) by reducing the restraint force constants. And,
eventually we just let system go for free simulations.
|
written
by Wonpil Im
|
|