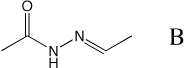
The initial
charmm script, gen_model_b.inp, generates B and
minimizes the structure in 4 different conformations. In addition, the script writes input files for the Gaussian
QM package that may be used to perform minimizations and frequency calculations
(gauss subdirectory). Several
inputs for the optimization of parameters associated with B are included.
water_model_b.inp: Calculates minimum interaction
energies and distances of water with B.
model_b_molvib.inp: Calculates the vibrational
spectra including performing normal mode assignments. The data in the resulting output are then compared with
results from a QM frequency analysis that may be done in CHARMM using the
script model_b_molvib_g03.inp.
model_b_surf_all_one.inp and model_b_surf_all_two.inp calculate several
potential energy surfaces for rotation around bonds in B. The two files are run consequtively to
obtain surfaces that are offset to zero; this could be performed in one file if
so desired. The resulting surfaces
may be compared to QM surfaces (see *.map files that include the basis set name
mp2_631gs).
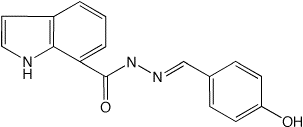
For the full drug
compound, gen_drug.inp, is used to generate and
minimize the compound and, model_2_surf_all.inp,
calculates two potential energy surfaces for comparison with QM data (see
*hf_631gs.map).
Subdirectory
gauss: The subdirectory contains a
number of input files for the Gaussian program including those created by
gen_model_b.inp and gen_drug.inp. See the 00readme file in that directory for
more information.