This tutorial illustrates how to use the replica exchange (REX)
molecular dynamics (MD) simulation together with the GBSW implicit
solvent to refine NMR structures. The simulation is carried out
by CHARMM and the MMTSB Tool Set. All required files including
this page can be downloaded
here.
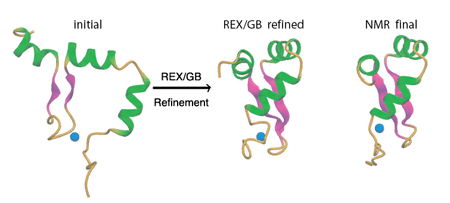
1. Iniitial Structures and NOE restraints
We will use the Zinc-dependent Redox Switch Domain of the
Chaperone Hsp33. This protein domain has a novel fold and the NMR
structure was solved by Jane Dyson's group at Scripps
and is now available from the
Protein Data Bank
with the PDB code
1XJH.
A minimized average structure of the NMR ensemble (
hsp.ref.pdb) can be viewed in VMD.
During the early stages of structure determination, only a limited
set of NOE distance restraints were assigned (see
mrdata/manual.mr), and the initial structure calcuation with
CYANA and CNS failed to identify the tertiary fold. The
representative structures computed by CNS are included in
inipdb/*.pdb. One can plot the residue NOE contacts
using gnuplot using commands,
gnuplot
set grid
plot 'data/mrdata/manual.mr' u 4:11 w p
The NOE data have already been translated to CHARMM format using
script
mr2charmm.pl, resulting in files
files/mrdata/manual.noe.*. They are grouped into intra-residue
(i=j), sequential (|i-j|=1), medium-range (|i-j|<=4) and long-range
(|i-j|)>4) NOEs. In this data set, there were only 30 long-range
NOEs, six of which define zinc coordination and the rest of which
are between residues in the beta-hairpin. There is virturally no
information about the tertiary fold and this is why the CYANA and
CNS calculations failed to converge.
2. REX/GB Refinement Simulation
The REX/GB refinement simulation requires both the initial
structures and the NOE distance restraints. The simulation is done
by script
rex.sge, which is also shown
below. The simulation length is 500 ps (1000 x 0.5 ps) and it
should take about 12 hours on the CTBP cluster with 2.40GHz Xeon CPU.
#!/bin/csh -f
# request the job to run at current director
#$ -cwd
# number of cpus
#$ -pe mpi 8
# walltime of the jobs (maximum is 48 hours)
#$ -l h_rt=12:00:00
#$ -N nmr_ref
#$ -e nmr_ref.err
#$ -o nmr_ref.log
#$ -m e
cat $TMPDIR/machines | sort -u | awk '{print $1" 2 /tmp/" user}' \
user=$USER > host.$$
aarex.pl -mp -hosts host.$$ -n 1000 -dir rexgb \
-log rexserver.log -charmmlog rex.log \
-mdpar param=22x,xtop=top22_zinc.inp,xpar=par22_zinc.inp \
-mdpar dynsteps=250,dyntstep=0.002,cuton=16,cutoff=16,cutnb=20 \
-mdpar gb=gbsw,gbswsgamma=0.01,gbswdgp=1.5,scalerad=nina,cmap=1 \
-par archive,natpdb=data/hsp.ref.pdb \
-custom setup data/noe.str -temp 16:300:600 data/inipdb/*.pdb
exit
This script differs from typical REX-MD simulations (such as de
novo peptide folding) in several places.
1). There are muliple initial structures.
aarex.pl will
automatically assign each initial structure to a replica. In
principle, one can also refine a single structure. However, in case
of NMR structure calcuation, it is better to use a diverse ensemble
of structures generated by CNS or DYANA, such that there is a
higher chance of sampling the true native fold.
2). The NOE restraints are imposed during the REX/GB simulation
through
-custom setup noe.str
option.
aarex.pl will read the script
noe.str before the dynamics runs for
all replicas. The purpose here is to combine experimental data with
modern GB force field to achieve better structural convergence than
either GB or NMR alone. The file
noe.str
is a simple CHARMM script that set up all the NOE distance
restraints.
3). Non-standard topology and parameter files are specified through
-xtop, -xpar options to handle the zinc
ion that coordinates with four cysteine residues.
3. Extracting, Clusterinng and Energy Minimization
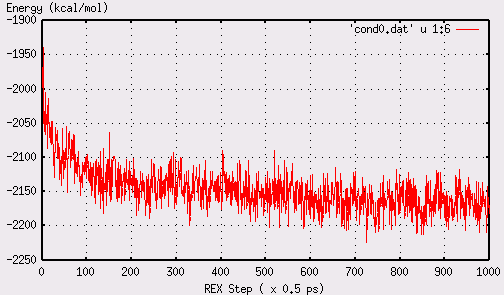
During the course of the simulation, one can examine the
convergence of energy using,
rexinfo.pl -bycond 0 -dir rexgb
It is also a good idea to make sure that exchange acceptance ratio
is sufficiently high, which is given in file
rexgb/rexserver.ninx. When the job is
finished, one can extract the structures sampled at the lowest
temperature and cluster them to obtain refined models. Results of
a previous run are included here (see data/rexgb_example). The
energy converges after 600-700 REX steps, shown below:
1).Run following command under
rexgb
directory to extract the last 200 structures at the lowest ensemble,
cd rexgb
../scripts/extractEns.pl -inx 801:1000
2). Go to the directory
ens0 that was
just created, and cluster the structures using
enscluster.pl with default options,
cd ens0
enscluster.pl -l 7:62 rex
The results of clustering can be checked by,
showcluster.pl rex
3). Compute the average structures of each clusters and minimize
them,
../../scripts/separate_cluster.pl rex
$CHARMMEXEC nfile=[NFILE] cid=[Cluster ID] < ../../scripts/cluster_ana.inp
[repeat for each clusters]
Scripts
extractEns.pl, separate_cluster.pl and
cluster_ana.inp are included in the
TAR/GZ file.

