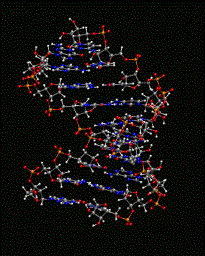
One can also use the MMTSB Tool convpdb.pl to perform this initial task. The command:
convpdb.pl -segnames -model 1 -out param22 1RRR.pdb > rr_1.pdb
produces a suitable pdb file with the needed translations. Small changes to the files
gen_rna_duplex.inp and gen_dna_duplex.inp will allow one to read the pdb file from
convpdb.pl. The needed changes are:
In original file replace:
open unit 20 read form name rr_1_noh.pdb.crd
read coor card unit 20
with:
rename segid n01a select segid rna1 end
rename segid n01b select segid rna2 end
open unit 20 read form name rr_1.pdb
read coor pdb unit 20 resid
rename segid rna1 n01a select segid n01a end
rename segid rna2 select segid n01b end
This renames the segment names for each chain to match those in the pdb file and then
changes them back so the segments have the same names in the later scripts.
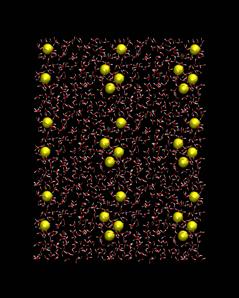
Once the duplex is generated waterbox to overlay the duplex is generated, . The box dimensions are determined by the dimension of the nucleic acid on which the calculation is being performed. Typically, the distance between the nucleic acid and the edge of the solvation cell is to be around 8-9 Å. In the present example script, watsodbox.inp, a water box with sodium ions (to neutralize the RNA) is created, with the resulting box shown below.