The objective of this tutorial is to
become familiar with the MMTSB Tool Set and learn how to prepare a system for
modeling tasks starting with a file from the Protein Data Bank. The goal
is to prepare a solvated system of a protein-RNA complex that can be used as the input for
simulation studies.
|
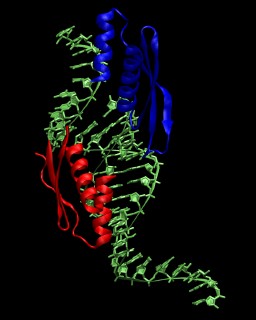
In this tutorial the double-stranded RNA-binding protein
Xlrbpa from Xenopus laevis is used as an example. A crystal structure for the
protein-RNA complex is available from the Protein Data Bank
with the PDB code
1DI2. A picture of the asymmetric unit is shown on the right. More
information about the crystal structure is available from this paper.
The system consists of two RNA-binding domains (chain A and B), four chains
of RNA (C, D, E, G), and the oxygen positions of 359 crystallographic water molecules.
The RNA in the biological complex is obtained by using the symmetry copy of chain E
instead of chain G. The first RNA strand consists of chain E followed by chain C, the
second RNA strand of chain D followed by the symmetry copy of chain E. According to
the PDB entry, the crystal structure was solved for a mutated protein (N112M).
|
|
| |
|
1. Obtain/copy the PDB file 1DI2.pdb from the Protein Data Bank to the current
working directory.
|
|
2. Extract the protein chains A and B
convpdb.pl -chain A 1DI2.pdb > 1di2.proteinA.pdb
convpdb.pl -chain B 1DI2.pdb > 1di2.proteinB.pdb
|
|
3. Reverse the N112M mutation to obtain the biological sequence with mutate.pl. Because the
mutation script does not handle chain IDs well, the chain ID of the input structure is first removed
(set to blank) and later reset to A or B respectively. Note, how multiple commands can be combined through
pipes so that the output from one command is used as input for the next command.
convpdb.pl -setchain " " 1di2.proteinA.pdb | mutate.pl -seq 112:N | \
convpdb.pl -setchain A > 1di2.proteinA.mutated.pdb
Repeat for chain B.
|
|
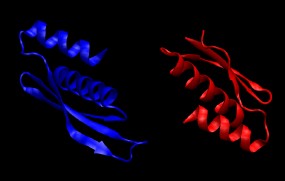
4. Combine the two protein chains into a single PDB file containing both chains.
convpdb.pl -merge 1di2.proteinB.mutated.pdb 1di2.proteinA.mutated.pdb > \
1di2.protein.pdb
Take a look at the resulting structure with VMD. It should look like this:
|
|
5. Extract the nucleic acid chains C, D, and E
convpdb.pl -chain C 1DI2.pdb > 1di2.rnaC.pdb
...
|
|
6. Generate the symmetry copy of chain E. The rotation matrix is available from the header
of the PDB file (see the second matrix under 'REMARK 350 APPLY THE FOLLOWING TO CHAINS: E').
convpdb.pl -rotate -1 0 0 0 1 0 0 0 -1 1di2.rnaE.pdb > 1di2.rnaE.sc.pdb
|
|
7. Generate the first RNA strand from chain E followed by chain C. This requires renumbering
of the chains from the original PDB.
convpdb.pl -setchain C -renumber 1 1di2.rnaE.pdb > \
1di2.rna.strand1.part1.pdb
convpdb.pl -renumber 11 1di2.rnaC.pdb > 1di2.rna.strand1.part2.pdb
Merge the two parts into a single file:
convpdb.pl -merge 1di2.rna.strand1.part2.pdb \
1di2.rna.strand1.part1.pdb > 1di2.rna.strand1.pdb
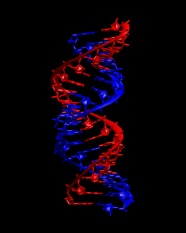
Repeat to generate strand 2 from chain D followed by the symmetry copy of chain E.
Merge the two strands into a single file:
convpdb.pl -merge 1di2.rna.strand2.pdb 1di2.rna.strand1.pdb > \
1di2.rna.pdb
Take a look at the resulting structure with VMD. It should look like this:
|
|
8. If you took a closer look at the resulting RNA duplex you might have noticed that the two
strands appear to be broken in the middle. This is an artifact of how the crystal structure
was obtained. The strand breaks can be "repaired" with a very short minimization in CHARMM because
CHARMM can automatically add missing atoms. The following command carries out only 10 steps of
steepest descent minimization. The 'nodeoxy' flag is needed to tell CHARMM that we are
working with RNA instead of DNA.
minCHARMM.pl -par nodeoxy,minsteps=0,sdsteps=10 1di2.rna.pdb > \
1di2.rna.fixed.pdb
Take a look at the resulting structure. The strand breaks should be gone. Also, the structure
now has all of the hydrogens compared to the PDB structure where hydrogens are missing because
they are difficult to resolve experimentally.
|
|
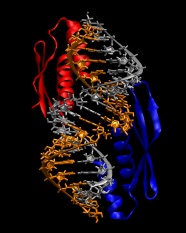
9. We are now ready to combine the protein and RNA into a single file:
convpdb.pl -merge 1di2.rna.fixed.pdb 1di2.protein.pdb > 1di2.complex.pdb
The model built so far should look like in the following image:
|
|
10. Next, we will add solvent to the complex. The crystal structure already
contains a number of water molecules. We will keep those and then add additional
waters around to fill a simulation box.
First, we extract the waters from the PDB file and add missing hydrogens. It is
convenient to keep the X-ray waters in a separate chain (chain X):
convpdb.pl -nsel water -renumber 1 1DI2.pdb | complete.pl | convpdb.pl \
-setchain X > 1di2.xray.waters.pdb
The X-ray waters are combined with the complex ...
convpdb.pl -merge 1di2.xray.waters.pdb 1di2.complex.pdb > \
1di2.complex.waters.pdb
... and then solvated in a cubic box with at least 9 Å between the complex or any
of the X-ray waters to the edge of the box:
convpdb.pl -solvate -cutoff 9 -cubic 1di2.complex.waters.pdb > \
1di2.complex.solvated.pdb
|
|
11. As a last step we need to add counterions to neutralize the system. The charge of
the protein-DNA complex can be obtained from CHARMM with the following command. Again,
the 'nodeoxy' option is used because the complex contains RNA instead of DNA.
enerCHARMM.pl -par nodeoxy -charge 1di2.complex.pdb
Counterions are added to solvated system by specifying the number of positive (SOD) and/or
negative ions (CLA). Decide how many and which type of ions we need to neutralize this
system from the output of the previous command and then run the following command:
convpdb.pl -ions <type>:<num> 1di2.complex.solvated.pdb > 1di2.complex.solvions.pdb
You can check whether the resulting system is neutral with:
enerCHARMM.pl -par nodeoxy -charge 1di2.complex.solvions.pdb
Finally, we should have a fully solvated system that is ready as a starting point for simulations.

|

