Specific directions:
1) Copy
these files into your working directory, unzip the files and extract the gaussian files
from gauss.tar.
2)
Evaluating lowest-energy conformations
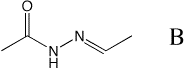
Generate the fragment "B" and minimize the structure in 4 different conformations:
$CHARMMEXEC < gen_model_b.inp > gen_model_b.out
Gaussian input files are created for each of these 4 conformations and are saved in the
gauss directory.
These files can be submitted to Gaussian within the gauss directory using the command:
g03 < model_b_opt_freq_mp2.com > model_b_opt_freq_mp2.g03out
(however, running Gaussian is beyond the immediate scope of the MMTSB Workshop tutorials)
3)
Evaluating hydration energies and distances
Calculate minimum interaction energies and distances of water molecules to fragment "B":
$CHARMMEXEC < water_model_b.inp > water_model_b.out
Four PDB files are generated mod1_wat1.pdb through mod1_wat4.pdb corresponding to the optimal positions of water molecules at different hydration sites on the fragment.
mod1_wat1.pdb .... O2...HOH
mod1_wat2.pdb .... N3-H3...OHH OUT OF PLANE
mod1_wat3.pdb .... N4...HOH OUT OF PLANE
mod1_wat4.pdb .... C5-H5...OHH OUT OF PLANE
The file model_b_wat.ene contains a summary of the interaction energies and distances of each of these structures along with the ab initio results. This information can be used to evaluate the quality of the existing CHARMM parameters for fragment "B" and decide whether better agreement is desired.
4)
Evaluating vibrational spectra
Calculate the vibrational spectra of fragment "B":
$CHARMMEXEC < model_b_molvib.inp > model_b_molvib.out
Compare the results with the QM frequencies (QM force constants stored in model_b_freq_g03.sec).
First, copy model_b_freq_g03.sec to fort.12:
cp model_b_freq_g03.sec fort.12
$CHARMMEXEC < model_b_molvib_g03.inp > model_b_molvib_g03.out
5)
Evaluating the conformational energy barriers
Calculate several potential energy surfaces for rotations around bonds in fragment "B":
$CHARMMEXEC < model_b_surf_all_one.inp > model_b_surf_all_one.out
Minumum energy structures for each surface are written to:
mod1_surf1_final.crd
mod1_surf2_final.crd
mod1_surf3_final.crd
$CHARMMEXEC < model_b_surf_all_two.inp > model_b_surf_all_two.out
The lowest-energy structure, model_b_min.crd, is used to properly zero the energy of the surfaces in the following .map files:
model_b_surf1.map
model_b_surf1.map
model_b_surf1.map
The information from these files can then be compared with the QM surfaces obtained from running Gaussian03 files (named: m1_s?_*.com) in the
gauss/surfaces directory. Note: Actually, the 00Readme file has an example input file that will "scan" through the desired dihedral angles. This can also be adapted for evaluating bond lengths, angles etc.
6)
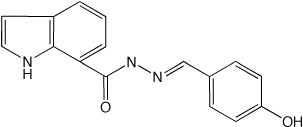
Evaluating lowest-energy conformations for the full drug molecule
The lowest-energy conformation and two potential energy surfaces for the full molecule can be obtained by:
$CHARMMEXEC < gen_drug.inp > gen_drug.out
$CHARMMEXEC < model_2_surf_all.inp > model_2_surf_all.out
As in step 2, drug_1_min.pdb, the lowest-energy conformation can be compared with that from ab initio calculations by using drug_1_opt_freq_mp2.com as the input file for Gaussian03.
As in step 5, model_2_surf1.map and model_2_surf2.map can be compared with potential energy surfaces generated from input files (named: m2_s*_*.com) in the
gauss/surfaces directory. Though, again, the "scanning" option in Gaussian, as described in gauss/surfaces/00Readme is a better way to go.