Objective and Overview
The objective of this tutorial is to introduce users to simple manipulations with
CHARMM and the MMTSB Tool Set. In this tutorial we will:
- Extract the 13-residue N-terminal peptide (C-peptide) from the enzyme
ribonuclease A (1RNU.pdb) using the MMTSB Tool Set tool
convpdb.pl.
- Visualize the peptide using vmd
- Go to the
VMD Tutorial to familiarize yourself with using VMD. Now display the pdb file of RNAase A
and highlight the peptide as shown in the figure at the right.
- Minimize the peptide using CHARMM and the script.
- Minimize the peptide using the MMTSB tool minCHARMM.pl.
- Compare the conformations and energies from the two calculations using
enerCHARMM.pl and rms.pl.
- Solvate the peptide using convpdb.pl.
- Minimize the solvated peptide w/ periodic (cubic) boundary conditions to prepare it for
molecular dynamics.
- Run 2 ps of molecular dynamics of the solvated peptide using the MMTSB Tool
mdCHARMM.pl
|
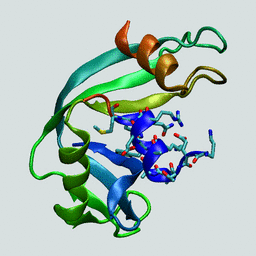
Structure of bovine ribonuclease A.
The N-terminal "C-peptide" is highlighted in blue.
|
We demonstrate the application of both CHARMM language scripting
and "shortcuts" utilizing the MMTSB Tool Set to carry-out more routine and fixed tasks.
We will show that, using the same parameters for both arguments to the MMTSB Tools and the CHARMM input
script, the same results emerge. One can use the MMTSB Tools to "bootstrap" your learning of the CHARMM
scripting language, and to provide basic templates for more complex tasks that require "programs" in the
CHARMM scripting language.
|
Extracting the N-terminal peptide and minimizing it
We must first extract the N-terminal peptide from the pdb file 1RNU.pdb that
you just downloaded. We will do this using the MMTSB Tool
convpdb.pl. During
the course of extracting the peptide from the pdb file we want to (i) "convert" from generic pdb residue
naming convention to the "charmm22" convention for atoms and residues, (ii) center the peptide, (iii)
eliminate any hetero atoms (HOH, ions, etc), (iv) extract residues 1 to 13 and (v) add a segment identifier
(segid) to the peptide segment. The following command accomplishes this:
convpdb.pl -center -sel 1:13 -segnames -out charmm22 \
-nohetero 1RNU.pdb > 1rnu_cpep.pdb
Note that the output has been directed to the file 1rnu_cpep.pdb
(you can download this file here if you are having troubles). Visualize the structure using vmd
to ensure you have what you want.
Now generate the components to carry-out molecular modeling on this structure by using CHARMM. This is
done by the script included below (download script rnase_cpep.inp).
The script first reads the necessary topology and parameter files into CHARMM. These are required to
establish the molecular building blocks and to describe the interactions between these blocks. In
"CHARMM-parlance" these files are used to generate the psf (protein structure file) for this peptide. The
psf contains all the pertinent information regarding the specific of bonding connectivity, non-bonded
exclusions, charges and other details necessary to manipulate the structure and compute its energy using
the CHARMM empirical force field. Look through the script below, the comments (statements preceded by a !)
provide additional explanations of the wheres, what's and whys. This script illustrates a number of the
"simple" CHARMM commands, including read/write, generate, internal coordinate (ic) build, harmonic positional
restraints, energy minimization using steepest descents (sd), root-mean-square distance (rmsd) calculation,
etc.
The input file for this example is given below:
|
* This tutorial example illustrates how to set-up and build
* a peptide in CHARMM.
* Note first we will "regularize" the peptide and then solvate it
* using the MMTSB Tool Set tool convpdb.pl
* In an accompanying example we will do all this with three mmtsb commands.
*
! Read in the parameter and topology files.
! We'll use the $CHARMMDATADIR as our source for top/param files
open unit 1 read form name "$CHARMMDATA/top_all22_prot_cmap.inp"
read rtf card unit 1
close unit 1
open unit 1 read form name "$CHARMMDATA/par_all22_prot_cmap.inp"
read param card unit 1
close unit 1
! We can read the sequence from the pdb file we created
open unit 1 read form name 1rnu_cpep.pdb
read sequ pdb unit 1
! Or we can add it explicitly - here we use the former
!read sequ card unit 5
!* Sequence for 13 residue N-terminal fragment of RNAse A
!* C-peptide
!*
!13
!LYS GLU THR ALA ALA ALA LYS PHE GLU ARG GLN HSD MET
generate pro0 first none last ct3 setup
rewind unit 1
read coor pdb unit 1 resi ! Read coordinates from pdb file using resid field
close unit 1
! Since the truncated peptide will not have the appropriate blocking groups,
! we add them here. Use ic build and then hbuild.
ic param
ic build
coor init select hydrogen end
! For consistency with defaults in the mmtsb tool minCHARMM.pl, we use
! this hbuild set-up
hbuild atom cdie eps 80.0 cutnb 10.0 ctofnb 7.5 ctonnb 6.5 shift vshift bygr
! Now we will do a rough minimization to "regularize" the structure and to
! remove any bad contacts from crystal structure prior to solvating the peptide
! We will harmonically restrain all peptide heavy atoms to ensure that
! the conformation does not change significantly from its initial state.
! Copy current coordinates to comparison set to see the motion.
coor copy compare
constrain harmonic force 5 mass select .not. hydrogen end
mini sd nstep 100 rdie switch vswitch cutnb 12 ctonnb 8 ctofnb 11 inbfrq -1
coor orie rms select .not. hydrogen end
open unit 1 write form name 1rnu_cpep_min.pdb
write coor pdb unit 1
stop
|
The peptide is minimized using CHARMM with the command:
$CHARMMEXEC < rnase_cpep.inp > rnase_cpep.out
The shell variable $CHARMMEXEC points to the CHARMM executable (it should be part of your shell set-up for the MMTSB Tool Set), the files produced are 1rnu_cpep_min.pdb and 1rnu_cpep.out. Take a look at the output file (using more or less), and look at what the output of the specific commands were. It is always essential that you look through your output and make sure there are not things you don't understand! Also use vmd to look at both the starting and final (minimized) structures. How do they differ?
We now carry-out the same task with the MMTSB Tool minCHARMM.pl. Note the correspondence between the arguments to this command and the arguments to various commands in the input script above.
minCHARMM.pl -par minsteps=0,sdsteps=100,sdstepsz=0.02 \
-par trunc=switch,dielec=rdie,cutnb=12,cuton=8,cutoff=11 \
-par param=22x \
-par xtop=top_all22_prot_cmap.inp \
-par xpar=par_all22_prot_cmap.inp \
-par blocked,nter=none,cter=ct3 \
-cons heavy self 1:13_5 -log mmtsbmin.log \
1rnu_cpep.pdb > 1rnu_cpep_mmtsbmin.pdb
Now calculate the energies of each final structure and compare using the MMTSB Tool enerCHARMM.pl:
First the mmtsb created conformation
enerCHARMM.pl -par param=22x \
-par xtop=top_all22_prot_cmap.inp \
-par xpar=par_all22_prot_cmap.inp \
-par trunc=switch,dielec=rdie,cutnb=12,cuton=8,cutoff=11 \
-par blocked,nter=none,cter=ct3 \
1rnu_cpep_mmtsbmin.pdb
Next the CHARMM script created conformation
enerCHARMM.pl -par param=22x \
-par trunc=switch,dielec=rdie,cutnb=12,cuton=8,cutoff=11 \
-par xtop=top_all22_prot_cmap.inp \
-par xpar=par_all22_prot_cmap.inp \
-par blocked,nter=none,cter=ct3 \
1rnu_cpep_min.pdb
Check the rmsd between the two structures using the MMTSB Tool rms.pl:
rms.pl 1rnu_cpep_min.pdb 1rnu_cpep_mmtsbmin.pdb
What is the rmsd between the two structures?
|
Adding solvation
In this section of the tutorial, we will use the MMTSB Tools to solvate our minimized peptide. First we'll add solvent and then we'll minimize the complete system with harmonic restraints on the peptide. The system will then be ready for molecular dynamics runs.
Add solvent and minimize, we do this all in two steps using convpdb.pl and minCHARMM.pl. First use convpdb.pl to solvate and add segment names - extract boxsize!
convpdb.pl -out charmm22 -solvate -cubic \
-cutoff 6 1rnu_cpep_mmtsbmin.pdb | \
convpdb.pl -out charmm22 \
-segnames > 1rnu_cpep_solv.pdb
We see that boxsize is a 34.743553 Å cube.
|
Now visualize the resulting solvated peptide box. It should look something like the figure below.
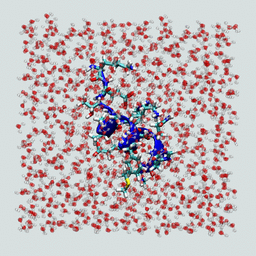
Does your solvated peptide look like this?
|
Now minimize with restraints and SHAKE constraints using PME electrostatics. Explore the minCHARMM.pl documentation a bit to understand the command below.
minCHARMM.pl -par minsteps=0,sdsteps=200,sdstepsz=0.02 \
-par trunc=switch,cutnb=12,cuton=8,cutoff=11 \
-par param=22x \
-par xtop=top_all22_prot_cmap.inp \
-par xpar=par_all22_prot_cmap.inp \
-par blocked,nter=none,cter=ct3 \
-par shake,boxsize=34.743553,nblisttype=bycb \
-cons heavy self 1:13_5 \
-cmd mmtsbminsolvate.inp -log mmtsbminsolvate.log \
1rnu_cpep_solv.pdb > 1rnu_cpep_solvmin.pdb
|
Suggested exercise:
In the previous commands, the MMTSB Tool Set has written the CHARMM input scripts with the -cmd option.
Use the saved command script (mmtsbminsolvate.inp) that the MMTSB Tool minCHARMM.pl created to run the calculation directly using CHARMM. Compare the automatically generated script to rnase_cpep.inp.
|
Running dynamics on this peptide in water using the MMTSB Tool mdCHARMM.pl
In this section we will take the configuration we just generated and run 2 ps of molecular dynamics on this system. We will, as we did in the minimization command above, employ periodic boundary conditions with Particle Mesh Ewald (PME) to account for the long-range electrostatics. Also, we run 1000 steps of molecular dynamics at 298 K in a constant pressure (NPT) ensemble. The pressure is maintained via the CPT algorithm and the temperature via the Nosé temperature bath. We save the energy output, the trajectory file (coordinates of the system versus time, in a compact binary format) and the final configuration. Explore the mdCHARMM.pl documentation a bit to understand the command below.
mdCHARMM.pl -par dynsteps=1000,dynens=NPT,dynitemp=298,dyneqfrq=1000 \
-par dynnose=1,dynoutfrq=10,dynpress=1,echeck=500 \
-par trunc=switch,cutnb=12,cuton=8,cutoff=11 \
-par param=22x \
-par xtop=top_all22_prot_cmap.inp \
-par xpar=par_all22_prot_cmap.inp \
-par blocked,nter=none,cter=ct3 \
-par shake,boxsize=34.743553,nblisttype=bycb \
-cmd mmtsbdynsolvate.inp -log mmtsbdynsolvate.log \
-enerout 1rnu_cpep_d0.ene -trajout 1rnu_cpep_d0.dcd \
-restout 1rnu_cpep_d0.res -final 1rnu_cpep_d0.pdb \
1rnu_cpep_solvmin.pdb
After running the dynamics (it should take about 5 minutes on a 3 GHz iMac), visualize the trajectory with vmd. To look at molecular dynamics trajectories with vmd, you need to read in a pdb and a dcd file. The following command will accomplish this.
vmd 1rnu_cpep_solvmin.pdb 1rnu_cpep_d0.dcd
|

|
In exploring this energy and temperature data, we see from the figure that the energy decreases as the system evolves in time, both the total and potential energy values evolve.
Similarly, the temperature initially rises and then begins to level off. In a more extended simulaiton, we would monitor such properties to identify when they plateau, indicating some level of "convergence".
|
We can get the energy and temperature data from 1rnu_cpep_d0.ene or from the output file, mmtsbdynsolvate.log, using the command:
grep "DYNA>" mmtsbdynsolvate.log
2nd column: Time(ps), 3rd column: Total Energy (kcal/mol), 4th column: Kinetic Energy, 5th column: Potential Energy and 6th column: Temperature (K).
|
|

