Tutorial and Examples for the CHARMM MM Workshop
Example input files and purpose:
- General tutorial and overview
- tutorial.inp
- tutorial2.inp (download these files)
Purpose: Illustrate a few CHARMM fundamentals
Usage:
charmm < input/tutorial.inp > output/tutorial.out
charmm < input/tutorial2.inp > output/tutorial2.out
Required files: none
- Initial conditioning and solvation of protein structure
- protein-generate_solvate.inp (download this example)
Purpose: Provide an initial set-up of protein psf (with or without
solvation)
Usage:
charmm pdbfile=1spz solvate=true crystal=true shape=octahedral
{toppar=$toppar seqname=1spz psf=false cutoff=9} <
input/protein-generate_solvate.inp > output/protein-generate.out
Required files: (any pair of)
top_all22_prot.inp, par_all22_prot.inp
toph19.inp, param19.inp,
top_all27_prot_na.rtf, par_all27_prot_na.prm
rtfparam.stream
and
@pdbfile.pdb
Where:
{...} means these are default values and don't need to be specified.
!* toppar ($toppar) ==> path to parameter and topology files
!**
!* pdbfile (...) ==> name of pdb coordinate file containing
!** coordinates and sequence information
!** (for psf=true key this is the name of the
!** coordinate file in pdb format (pdbfile_minimized.pdb)
!** and the pre-generated psf file (pdbfile.psf))
!* version (22) ==> default version number for rtf and parameters
!** choices 22, 19 or 0 if version = 0 then rtf
!** and parameters read from file rtfparam.stream
!* psf (false) ==> key specifying use of pre-generated psf and
!** pdb coordinate file for use w/ multi-segid
!** solute systems
!* seqname (pdbfile) ==> name of protein sequence (segid)
!**
!* shape (cubic) ==> variable specifying whether truncated octahedral or
!** cubic volume used (octahedral/cubic)
!* crystal (false) ==> logical variable true/false specifying whether
!** to use crystal orientation of simulation volume
!* cutoff (9) ==> value of cutoff to be used in establishing how
!** far images are to be from volume "edges", often set
!** at or near nonbonded list cutoff value
!** to use crystal orientation of simulation volume
!* solvate (false) ==> logical flag which determines whether you stop after
!** initial minimization and psf generation or proceed
- Initial conditioning and solvation of DNA structure
- DNA-ions-n-solvate.inp (download this example)
Purpose: Provide an initial set-up of DNA + Na+ counter ions psf (with or without solvation)
Usage:
charmm pdbfile=103d solvate=true crystal=true shape=cubic
{toppar=$toppar seqname=103d psf=false cutoff=9} <
input/DNA-ions-n-solvate.inp > output/DNA-ions-n-solvate.out
Required files: $toppar/top_all27_na.rtf, $toppar/par_all22_na.prm, @pdbfile.pdb
Where: {...} means these are default values and don't need to be specified.
!* toppar ($toppar) ==> path to parameter and topology files
!**
!* pdbfile (...) ==> name of pdb coordinate file containing
!** coordinates and sequence information
!** (for psf=true key this is the name of the
!** coordinate file in pdb format (pdbfile_minimized.pdb)
!** and the pre-generated psf file (pdbfile.psf))
!* psf (false) ==> key specifying use of pre-generated psf and
!** pdb coordinate file for use w/ multi-segid
!** solute systems
!* shape (cubic) ==> variable specifying whether truncated octahedral or
!** cubic volume used (octahedral/cubic)
!* crystal (false) ==> logical variable true/false specifying whether
!** to use crystal orientation of simulation volume
!* cutoff (9) ==> value of cutoff to be used in establishing how
!** far images are to be from volume "edges", often set
!** at or near nonbonded list cutoff value
!** to use crystal orientation of simulation volume
!* solvate (false) ==> logical flag which determines whether you stop after
!** initial minimization and psf generation or proceed
- Estimate of pKa values for protein residues using single site approximation with generalized Born and/or Poisson-Boltzmann continuum approximations.
- protein-pKa.inp (download this example)
Purpose: Calculate estimates of deltapKa values for protein or peptide
Usage:
charmm pdbfile=1hel minimize=false epsinterior=10 pb=false size=0.75
< input/protein-pKa.inp > output/protein-pKa.out (illustrate
GB pKa calculation for whole protein)
charmm pdbfile=phel minimize=false
epsinterior=10 pb=false size=0.75 < input/protein-pKa.inp >
output/protein-pKa_pb.out (illustrate GB & PB pKa
calculation for protein fragment)
Required files: $toppar/top_all22_prot.inp, $toppar/par_all22_prot.inp, @pdbfile.pdb
Where: {...} means these are default values and don't need to be specified.
!* toppar ($toppar) ==> path to parameter and topology files
!**
!* pdbfile (...) ==> name of pdb coordinate file containing
!** coordinates and sequence information
!**
!* minimize (false) ==> logical determining whether to condition
!** pdb structure by minimization or not
!**
!* epsinterior (10) ==> value to sue for protein interior dielectric
!** constant
!* pb (false) ==> logical indicating whether to use Poisson-Boltzmann
!** continuum model to do pKa calculation
!* size (0.75) ==> grid spacing to use in finite difference PB
!** calculation
- Free energy perturbation methods - mutation of protein residue (Y33F)
- protein_FEP-by-TSM.inp (download this example)
Purpose: Illustrate
calculation of free energy differences for perturbation of amino acid residue
in
protein and reference (peptide) state using CHARMM TSM (Thermodynamic
Simulation Modeule).
Usage:
charmm pdbfile=1pgb resnum=33 lambdastep=0.3 dynamics=true
analysis=true < input/protein_FEP-by-TSM.inp >
output/protein_FEP-by-TSM.out (perturbation in
protein)
charmm pdbfile=rpgb resnum=33 lambdastep=0.3 dynamics=true analysis=true < input/protein_FEP-by-TSM.inp > output/protein_FEP-by-TSM_ref.out (perturbation in reference peptide)
Required files: $toppar/top_all22_prot.inp, $toppar/par_all22_prot.inp, @pdbfile.pdb
Where: {...} means these are default values and don't need to be specified.
!* toppar ($toppar) ==> path to parameter and topology files
!**
!* pdbfile (...) ==> name of pdb coordinate file containing
!** coordinates and sequence information
!** (for psf=true key this is the name of the
!** coordinate file in pdb format (pdbfile.pdb)
!** and the pre-generated psf file (pdbfile.psf))
!* psf (false) ==> key specifying use of pre-generated psf and
!** pdb coordinate file for use w/ multi-segid
!** solute systems
!* resnum (33) ==> residue number of mutation site
!* lambdastep (0.1) ==> stepsize for FEP perturbations
!* dynamics (false) ==> logical determining whether FEP
!** simulation to be performed
!* analysis (false) ==> logical determining whether analysis of FEP
!** simulation to be performed
- Results:


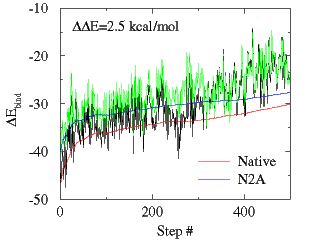
- Approximate association free energy for leucine zipper
- 2zta_bind-w-GB.inp (download this example)
Purpose: Illustrate the GB/MM approach to estimating free energy difference for two examples. 1) Association energy of leucine zipper dimer from analysis of native 2zta leucine zipper trajectory. 2) Association energy for Asn16Ala mutant of leucine zipper 2zta computed from same trajectory ("alanine scanning").
Usage:
charmm N2A=false pdbfile=1pgb < input/2zta_bind-w-GB.inp > output/2zta_bind-w-GB.out (association
in native leucine zipper)
charmm N2A=false pdbfile=1pgb < input/2zta_bind-w-GB.inp > output/2zta_bind-w-GB_N2A.out (association in mutant leucine zipper)
Required files: $toppar/toph19.inp,
$toppar/param19.inp, ./crd/2zta_d1-60p-sub.hex
- Results: