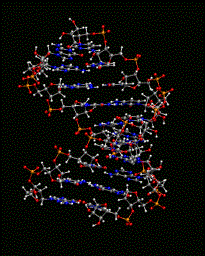
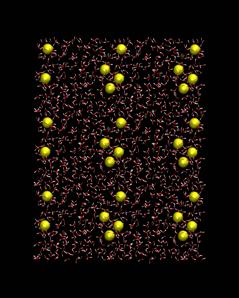
The remainder of the CHARMM scripts involve equilibration of the system around the RNA followed by the production MD simulation.
$CHARMMEXEC < equil.inp > equil.out &
During equilbration, equil.inp, the coordinates of the DNA/RNA are harmonically constrained and a minimization is performed. This is followed by a NVT simulation (minimum of 20 ps) to allow the solvent and ions to equilibrate around the nucleic acid. The final coordinates from the equilibration are used for the production simulation, dyn_npt_1.inp.
$CHARMMEXEC nstep=5000 < dyn_npt_1.inp > dyn_npt_1.out &
The production simulation involves an NPT simulation with long-range electrostatics treated via the Particle-Mesh Ewald method. Note that the trajectory is run in a loop such that a series of trajectory (.trj) files along with the associated restart and energy files are created. This allows the simulation to be readily restarted and facilitates handling of the files during analysis.
It is probably not feasible to run the production on your workstation in a short time. If you wish to try, you will need to reduce the nstep variable on the command line.
Two simple analysis CHARMM scripts to determine the RMS Differences as a function of time (rms.inp) and the base pairing energy as a function of time (bp_interactions.inp) are included in the analysis subdirectory.
Then, run the input files:
cd analysis
cp ../rms.inp .
cp ../bp_interactions.inp .
$CHARMMEXEC nstep=5000 < rms.inp > rms.out
$CHARMMEXEC nstep=5000 < bp_interactions.inp > bp_interactions.out